MagpiEM: Automating the cleaning of particles for sub-tomogram averaging
- Abstract number
- 572
- Presentation Form
- Poster
- DOI
- 10.22443/rms.mmc2023.572
- Corresponding Email
- [email protected]
- Session
- Poster Session Three
- Authors
- Mr Frank Nightingale (1)
- Affiliations
-
1. University of Oxford
- Keywords
Cryo-EM
Cryo-ET
Data processing
Sub-tomogram averaging
Lattice cleaning
- Abstract text
Cryo-electron tomography (Cryo-ET) provides an unprecedented look at biomolecules in their native state, whereby sub-tomogram averaging of molecules in cellular lamellae can give a fully native-state picture of a protein’s structure. This technique proves invaluable where isolated proteins do not provide an adequate picture, for example where the protein’s function is lost upon isolation1, or when studying a protein’s capacity to form macroscopic structures2. Cryo-ET also comes with challenges, however, due to the crowded and inhomogeneous nature of the native cellular environment. Sub-tomogram averaging requires accurate knowledge of the positions of proteins within tomograms, for which several approaches have been developed, including template-matching, random picking across membranes, and machine-learning based methods3. These methods must employ generous criteria to avoid missing particles, but this inevitably leads to many false-positive particles, often largely outnumbering the number of correct hits. Accordingly, particle data must be “cleaned” to remove these false positives. This process involves a large amount of tedious, manual work, with analysis of large datasets requiring weeks of manually clicking particles. This not only introduces a large amount of unavoidable human error, but often encourages the use of sub-optimal picking criteria to effectively manage this workload.
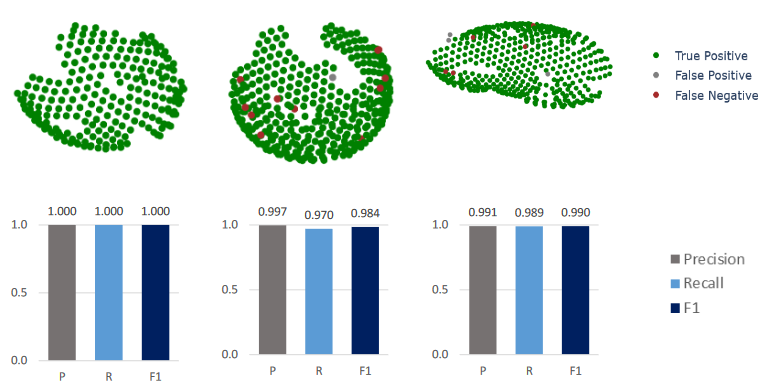
Thus, we have developed MagpiEM, a tool for automating the cleaning of these datasets. By computationally comparing geometric parameters between particles, assembling them into lattices with a given curvature, we can provide results indistinguishable from manual picking (Fig. 1), while decreasing the total time taken per dataset by upwards of 99%. We have evaluated this software on a variety of biological systems, including HIV Gag structural proteins in immature virions, phycobilisomes in cyanobacteria, Tsr lattices in E. coli, and particulate methane monooxygenase in methanotrophic bacteria. The software can also function as a library for general particle analysis and visualisation, for which we have provided examples including the visualisation of changes in particle position throughout refinement, and the comparison of nearest-neighbour angles between particles.
The software is open-source and available on github: https://github.com/fnight128/MagpiEM
Fig. 1: Precision and recall of MagpiEM vs manual picking of Gag lattice data
- References
1. Zhu, Y. et al. Structure and activity of particulate methane monooxygenase arrays in methanotrophs. Nat Commun 13, 5221 (2022).
2. Mendonça, L. et al. CryoET structures of immature HIV Gag reveal six-helix bundle. Commun Biology 4, 481 (2021).
3. Pyle, E. & Zanetti, G. Current data processing strategies for cryo-electron tomography and subtomogram averaging. Biochem J 478, 1827–1845 (2021).